网站首页生活常识 >正文
催化表面的精细结构对结构敏感反应具有显着影响,高通量(HT)筛选和机器学习(ML)被认为可以有效探索这些影响的潜在规律并加速催化剂的开发。然而,报道的机器学习框架过于粗糙,无法精确预测催化性能。
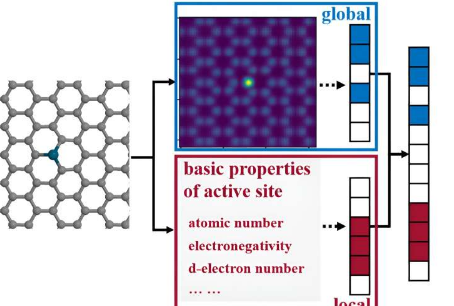
目前常用的两种转换方法是描述符和图。然而,描述符的构建通常会忽略原子连接,这使得机器学习模型很难捕获与催化性能最相关的详细几何信息。
基于图的机器学习模型在更新节点的过程中不可避免地会丢失吸附位点的几何排列信息,而消息传递神经网络的复杂性导致其对电子或几何结构不敏感,可解释性差。因此,仍然缺乏可解释的机器学习框架,可以同时捕获多相催化中电子和几何精细结构的特征中电子和几何精细结构的特征。
最近,中国浙江大学王勇教授领导的研究团队创建了一个名为GLCNN的数据增强卷积神经网络(CNN)机器学习框架,它结合了“全局+局部”的特征。该框架可以通过将催化表面和吸附位点分别转换为二维网格和一维描述符,无需复杂的编码方法即可捕获原始精细结构。
数据增强(DA)的加入可以扩展数据集,缓解化学数据集不足造成的过拟合。GLCNN框架准确预测并区分了OH在一组类似的碳基过渡金属单原子催化剂(TMSAC)上的吸附能,平均绝对误差(MAE)小于0.1eV,位居流行模型的最佳结果到目前为止已经在大型数据集上进行了训练。成果发表于催化学报上。
将GLCNN与基于描述符或基于图的模型进行比较,发现比较模型无法准确预测含有IB和IIB过渡金属或顺式/反式构型的催化剂的OH吸附能。GLCNN模型的预测性能明显优于对比模型,表明网格和描述符的结合可以更好地反映催化活性中心的电子和精细几何信息。
与传统的CNN和基于描述符的单侧特征提取不同,这种精细结构敏感的ML框架可以通过无偏的可解释性,从几何和化学/电子特征(例如对称性和配位元素)中提取影响催化性能的关键因素。分析。
对描述符部分的特征重要性分析表明,吸附位点的电子结构和对称性元素至关重要,且金属的重要性强于其配位环境。对各层的可视化分析表明,GLCNN能够自动提取符合人类直觉的化学结构的几何信息。
随着层数的加深,GLCNN逐渐寻求基于基础催化知识的特征提取方向,提取更抽象的有利于吸附能预测的高维特征。该框架为具有广阔物理和化学空间的多相催化剂的高精度高温筛选提供了可行的解决方案。
版权说明:本站所有作品图文均由用户自行上传分享,仅供网友学习交流。若您的权利被侵害,请联系我们
相关文章:
- 2023-11-26研究表明南极臭氧层空洞在仲春时更深
- 2023-11-24机器人假肢脚踝改善自然运动和稳定性
- 2023-11-24球形机器人来救援
- 2023-11-24解释人工智能的方法可能并不那么容易解释
- 2023-11-24描述开放系统中量子信息加扰的通用框架
- 2023-11-24研究为抗生素耐药性和健身景观提供了新的见解
- 2023-11-24物理学家发现量子材料中奇异电荷传输的证据
- 2023-11-23一种高效去除水产养殖废水中磷酸盐的方法
- 2023-11-23研究人员在防止钒电池容量损失方面获得了有希望的结果
- 2023-11-23了解化学处理沙土的强度发展机制
- 站长推荐
- 栏目推荐
- 阅读排行
- 健康和教育密切相关新西兰需要将其更多地融入小学
- Steam现已全面支持DualShock和DualSense控制器无需购买新的Xbox控制器
- DistrictTaco希望扩大其在罗利地区的业务
- Humane的AiPin–您的新型可穿戴人工智能助手
- Microsoft365CopilotAI如何提高您的工作效率
- MicrosoftRadius云开源应用程序平台
- 生产目的FiskerPear具有透明A柱因为移动头部太困难
- 索尼Xperia5V马来西亚发布Snapdragon8Gen2SoC 8GBRAM 256GB储存空间起价RM4999
- Nissan的模块化PulsarSportbak集轿跑车 旅行车和皮卡于一体
- 新奥尔良烤肉店将在中央市场推出