网站首页生活常识 >正文
将不同层次的概况整合在一起的多组学分析具有挑战性,因为概况中的变量数量彼此差异很大。例如,基因表达谱和基因组DNA甲基化谱经常一起分析;然而,基因只有数万个,而DNA甲基化位点却多达数千万个。
这些数字相差几个数量级,并且基因和DNA甲基化位点之间的配对数量非常巨大。因此,需要大量的计算资源,根据先验知识,通过关注启动子区域等特定区域的DNA甲基化位点,在不控制靶标数量的情况下进行综合分析。然而,由于限制了待分析的基因组区域,DNA甲基化对其他区域(例如增强子)和功能的影响仍未得到探索。
学习情况及成果
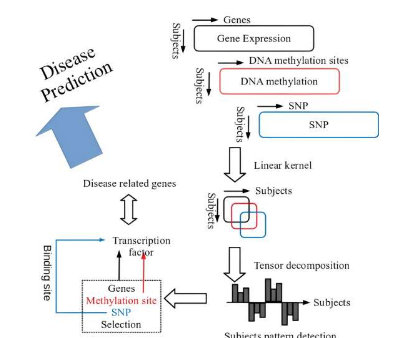
最近发表在PLOSONE杂志上的一项研究应用了先前研究中开发的方法来处理岩手东北医疗大银行组织(IMM)综合收集的多组学数据(基因表达谱、DNA甲基化谱、单核苷酸多态性(SNP)谱)对100名当地居民参与者进行了研究,并确认是否可以识别与疾病相关基因的关系。
这是一种数据驱动的方法,称为变量提取方法,采用基于核张量分解的无监督研究(以下称为张量分解),该方法适用于所有受试者都属于健康组的数据集。
此外,该方法可以通过每1个图谱的内核大小(特别是受试者参与者的平方)左右的记忆来实现,因此,甚至对于包含数千万个SNP或DNA甲基化位点的基因组和表观基因组等巨大的图谱也是如此。数据驱动的分析可以识别研究对象的独特模式,并识别与这些模式表现出相似性的变量(基因表达谱、DNA甲基化谱、SNP谱)。
在这项研究中,张量分解应用于从三种细胞类型(CD4阳性T细胞、单核细胞和中性粒细胞)检索的每种常染色体的多组学数据。结果,确定了受试者概况的两种模式,并且在22个常染色体中观察到的这两种受试者模式显示出其他常染色体之间非常强的相互相关性。由于每个常染色体中鉴定的基因彼此完全独立,这表明观察到的跨染色体共享的模式并不是巧合。
观察到的正交模式也不能用批次效应来解释,并且在通过不同方法获得的三个组学概况中不可能存在相同的批次效应。这两种模式的主体分别通过张量分解获得作为第二和第三奇异值向量。从所有三种细胞类型中检测到第二奇异值向量,而从除单核细胞之外的两种细胞类型中检测到第三奇异值向量。
然后,选择与这些模式具有同源性的基因和DNA甲基化区域,以发现这些基因和区域是许多转录因子的靶标。此外,富集分析表明这些转录因子与多种疾病相关。
此外,研究发现,所识别的SNP在统计上与这些转录因子的结合位点显着重叠。因此,作者认为张量分解的应用对于多组学数据集的集成分析是有效的。
版权说明:本站所有作品图文均由用户自行上传分享,仅供网友学习交流。若您的权利被侵害,请联系我们
相关文章:
- 2023-11-26研究表明南极臭氧层空洞在仲春时更深
- 2023-11-24机器人假肢脚踝改善自然运动和稳定性
- 2023-11-24球形机器人来救援
- 2023-11-24解释人工智能的方法可能并不那么容易解释
- 2023-11-24描述开放系统中量子信息加扰的通用框架
- 2023-11-24研究为抗生素耐药性和健身景观提供了新的见解
- 2023-11-24物理学家发现量子材料中奇异电荷传输的证据
- 2023-11-23一种高效去除水产养殖废水中磷酸盐的方法
- 2023-11-23研究人员在防止钒电池容量损失方面获得了有希望的结果
- 2023-11-23了解化学处理沙土的强度发展机制
- 站长推荐
- 栏目推荐
- 阅读排行
- 健康和教育密切相关新西兰需要将其更多地融入小学
- Steam现已全面支持DualShock和DualSense控制器无需购买新的Xbox控制器
- DistrictTaco希望扩大其在罗利地区的业务
- Humane的AiPin–您的新型可穿戴人工智能助手
- Microsoft365CopilotAI如何提高您的工作效率
- MicrosoftRadius云开源应用程序平台
- 生产目的FiskerPear具有透明A柱因为移动头部太困难
- 索尼Xperia5V马来西亚发布Snapdragon8Gen2SoC 8GBRAM 256GB储存空间起价RM4999
- Nissan的模块化PulsarSportbak集轿跑车 旅行车和皮卡于一体
- 新奥尔良烤肉店将在中央市场推出